Introduction
Sarcoidosis is an idiopathic, multisystemic inflammatory disease characterized by non-caseating granulomas. Although it can affect any organ system, it predominantly involves the lymph nodes, lungs, skin, and eyes [1].
Cell-mediated, granulomatous inflammation remains the pathological hallmark, but a thorough understanding of the pathogenesis remains unclear [2].
While there is no unanimous agreement on whether sarcoidosis is classified as an autoimmune disease, there is a growing body of evidence suggesting that certain forms of sarcoidosis might involve autoimmune factors. Pre-sently, our comprehension of the development of sarcoidosis is predominantly based on research into pulmonary sarcoidosis. The extent to which these findings apply to neurosarcoidosis, and whether the immune responses in neurosarcoidosis deviate from those seen in sarcoidosis affecting other bodily systems, are subjects undergoing active research [1,3].
Neurosarcoidosis usually affects women more than men, usually in middle age. The prevalence widely varies, with the highest reported incidence among Scandinavians and African Americans [1].
Neurological involvement occurs in about 5-10% of sarcoidosis. In about 30-70% of the cases, the presentation might be neurological; however, at the time of initial evaluation, the majority of individuals will have an extraneural disease. It can involve both the central nervous system (CNS) and the peripheral nervous system (PNS). Of the many possible manifestations of neurosarcoidosis, cranial nerve palsy is the most prevalent form, followed by meninges, ventricles, hypothalamo-pituitary axis (HPA), spinal cord, brain stem, and cerebellum [4,5]. Although rare, isolated neurosarcoidosis has also been reported. About 31% of cases have the systemic disease at the time of presentation, with 84% of cases eventually developing systemic manifestations [6].
Elevated angiotensin-converting enzyme (ACE), pleocytosis, IgG index, increased protein, and the presence of oligoclonal bands can all be identified in cerebrospinal fluid (CSF) analysis. Elevated serum ACE levels, hypercalcaemia, and elevated erythrocyte sedimentation rate could suggest sarcoidosis. Laboratory workup is often nonspecific and may only be used as an adjunct marker [7].
On imaging, neurosarcoidosis mimics many infectious, neoplastic, demyelinating, and inflammatory diseases and hence can be a difficult definitive diagnosis to make [4,7,8].
Positron emission tomography (PET)/computed tomography (CT) in neurosarcoidosis is primarily used to assess systemic sarcoidosis, determine suitable tissue biopsy sites, and assess response to treatment. The normal uptake of fluorodeoxyglucose (FDG) by brain parenchyma often impairs the assessment of neurological involvement [7,9].
Performing a neural tissue biopsy is infrequent due to the associated risks, which can lead to permanent neurological impairments. It is essential to prioritize the exploration of alternative targets such as endobronchial tissue or lymph nodes for biopsy. In all cases, obtaining tissue cultures is vital because conventional stains for detecting organisms like mycobacteria and fungi might not be dependable [3,4]. Nonetheless, when confronted with rapidly progressing and unconventional symptoms that cause significant neurological deterioration, the situation warrants a neural biopsy. Another pitfall that might arise is the histological identification of non-specific chronic inflammation, especially because patients within this subgroup are often initiated on high-dose steroids [7].
To improve the diagnosis and the management of patients with suspected neurosarcoidosis, the 2018 Neurosarcoidosis Consortium Consensus Group (NCCC) [10] recently recommended new diagnostic criteria. Based on the clinical features and histopathology, the diagnosis of neurosarcoidosis can be classified into the following:
possible – typical clinical and radiological features without histopathological diagnosis,
probable – histopathological confirmation of a systemic disease,
definitive – central nervous system biopsy consistent with sarcoidosis.
Peripheral nervous system involvement is also considered a part of neurosarcoidosis [10].
The diagnostic criteria for probable neurosarcoidosis, as outlined by the 2018 NCCC, contrast with those presented by Zajicek et al. [11]. In the latter, meeting the criteria involves the presence of oligoclonal bands, elevated levels of protein and/or cells in cerebrospinal fluid, indicators of systemic sarcoidosis such as histology, Kviem’s test, and serum ACE, along with 2 imaging markers (such as chest imaging or gallium scan), and the exclusion of other potential diagnoses [11].
Patients falling into the category of “possible neurosarcoidosis” should be approached with caution because more than half of them received alternate diagnoses on neural biopsy in a recent study [7].
Clinical presentation
Symptoms of neurosarcoidosis are often non-specific and depend on the site of involvement. Facial nerve palsy and vision loss are the most common presentations, followed by headache, seizures, weakness, paraesthesia, and meningeal signs. Additionally, myelopathy and polyneuropathy can be found [6,12]. Involvement of the HPA may lead to symptoms of diabetes insipidus (DI) [12]. Rarely, it can present acutely with stroke or haemorrhage [13].
Imaging findings
Meningeal involvement
Meningeal inflammation is one the most common manifestations of neurosarcoidosis, occurring in 16-69% of the patients. Leptomeningeal involvement is more common than pachymeningitis [14].
Leptomeningeal involvement occurs in about 40% of the cases, appearing as thickening and enhancement of the leptomeninges on post-contrast T1 images (Figure 1A). The thickening can either be nodular or diffuse [12]. A predilection for the basal meninges and perivascular spaces is seen, but it can occur anywhere across the neuro-axis [15]. These imaging appearances are practically indistinguishable from the leptomeningeal involvement seen in infections (tubercular and fungal), lymphoma, and primary angiitis of the CNS. Care must be exercised to differentiate leptomeningitis from normal blood vessels, haemorrhage, and melanin [8].
Figure 1
A) Sagittal T1 + C image shows nodular leptomeningeal enhancement in the parietal region (yellow arrow) and cerebellar hemispheres (white arrow). B) Coronal T1 + C image shows smoothly thickened tentorial leaflets bilaterally (red arrows). C) Axial T1 +C image shows enhancement of the meatal segments of bilateral seventh cranial nerves (green arrows). D) Axial CT of the skull shows a lytic lesion with no peripheral sclerosis involving the right parietal bone (blue arrow)
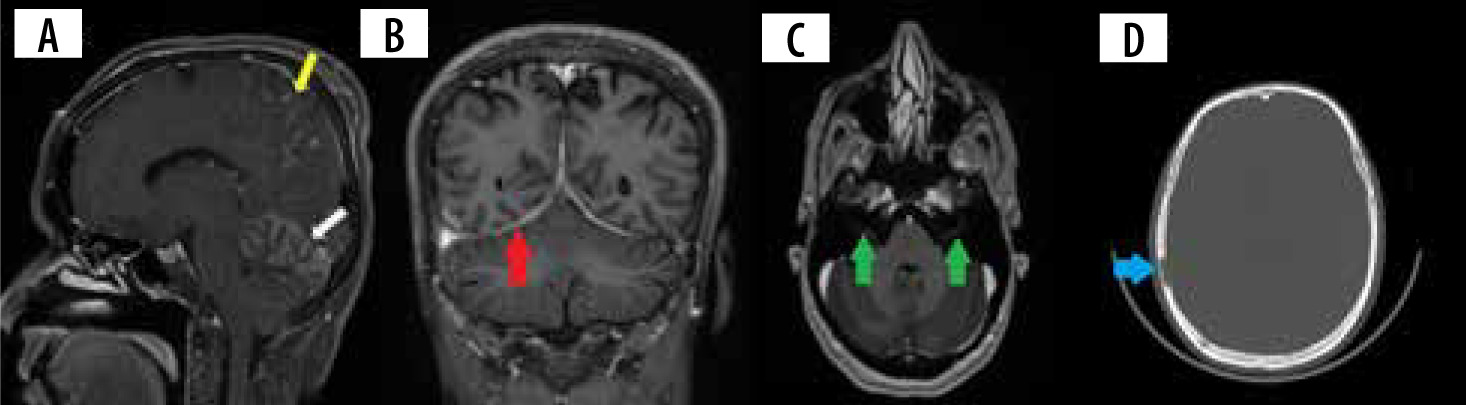
Pachymeningitis in neurosarcoidosis is predominantly found in the region of the central skull base involving the cavernous sinus, sphenoid wing, and clivus. The involvement can be single or multifocal, smooth or nodular, and can uncommonly demonstrate a dural tail (Figures 1B and 2B) [14]. Most of these lesions show homogenous contrast enhancement. They can often be T2 hypointense and can be confused with calcified meningiomas and metastases [12]. Larger nodular lesions can cause compressive cranial nerve palsies, parenchymal oedema, and subfalcine herniation. Because a wide range of neoplastic, infectious, and inflammatory disorders are included in the differential diagnosis of pachymeningitis, involvement of the leptomeninges, HPA, orbit, salivary glands, and brain parenchyma may suggest neurosarcoidosis. Nevertheless, tissue biopsy remains the gold standard for diagnosis in isolated pachymeningitis [14].
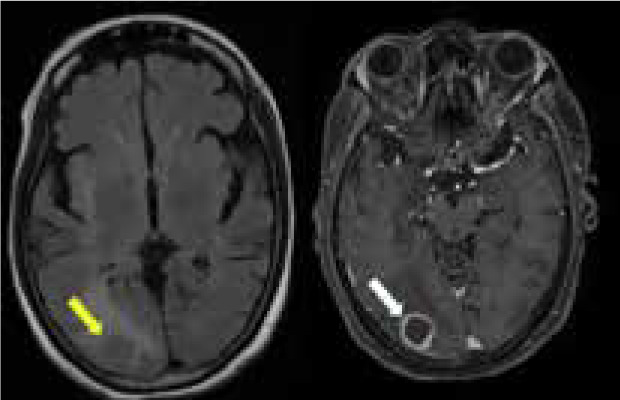
Parenchymal involvement
Non-enhancing white matter lesions are reported to be the most common parenchymal abnormality, occurring in about 56% of patients. These lesions are predominantly seen in the periventricular and white matter regions. The effect of age and the presence of concomitant vascular comorbidities can be a potential confounder because differentiating these 2 entities with neurosarcoidosis is not possible on imaging [16].
The granulomas in neurosarcoidosis can manifest as single or multifocal. These lesions show diffuse or rim enhancement, which is considered to be the centripetal involvement of the perivascular Virchow-Robin (VR) spaces. Often, they can demonstrate non-enhancing areas (Figure 3). Though necrosis and calcification are rare, they can be complicated by haemorrhage. Intra-axial masses appear isointense on T1 and hyperintense on T2. They can easily be mistaken for gliomas, lymphomas, haemorrhage, demyelinating disease, or metastasis, in which case leptomeningeal involvement favours neurosarcoidosis [12,16-18].
Cranial nerve involvement
Cranial nerve involvement is one of the common manifestations of neurosarcoidosis, but it can be challenging to diagnose. The optic nerve and facial nerve are commonly involved, identified on imaging as nerve thickening and enhancement. Leptomeningeal disease is often present. Optic nerve involvement can mimic optic neuritis or gliomas [12,18]. In cases of sellar involvement, there is often an extension that affects the chiasm as well. Bilateral engagement is more frequent, and unfortunately this often leads to unfavourable visual outcomes [3,19]. Ophthalmic examination often reveals concomitant uveitis, scleritis, and orbital masses [19].
Orbital apex lesions can cause compressive neuropathy of multiple upper cranial nerves [20]. Neurosarcoidosis is one of the important causes of bilateral facial nerve palsy along with Lyme disease and Guillain-Barré syndrome (Figure 1C) [8]. Differentials to consider include perineural spread of tumour, carcinomatosis, and lymphoma [18].
Hydrocephalus
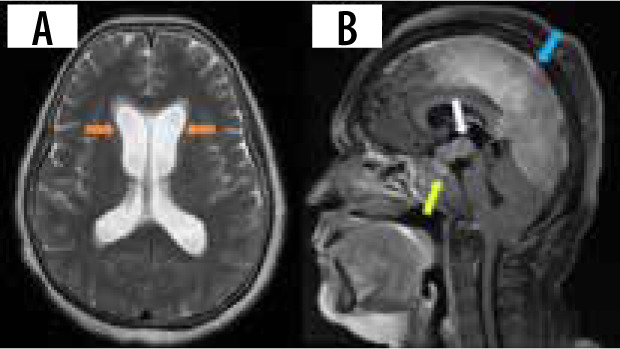
Hydrocephalus is another uncommon manifestation, occurring in about 4-38% of cases [16]. The communicating type is usually due to altered and decreased CSF resorption, and the obstructing type occurs due to adhesions from the leptomeningeal involvement. Magnetic resonance imaging (MRI) shows diffuse enlargement and ballooning of the ventricles with white matter changes and transependymal effusion of CSF (Figure 2A). Contrast-enhanced MRI can show linear or patchy enhancement within the ventricles, with concomitant meningeal enhancement. A wide range of potential differential diagnoses should be considered, encompassing post-traumatic, infectious, inflammatory, and neoplastic causes. This is particularly important when there is no evident history suggestive of neurosarcoidosis [21]. FLAIR MRI can show differential intensity in unilateral hydrocephalus due to elevated protein concentration in the involved ventricle [12].
Hypothalamo-pituitary axis involvement
Involvement of HPA can be seen in 9-17% of cases [7,22]. Leptomeningitis around the pituitary gland and hypothalamus can involve the HPA, presenting with clinical symptoms of hypopituitarism and DI. On post-contrast T1 images, thickening and enhancement of the pituitary stalk and hypothalamus can be seen. Pseudo adenoma, pseudo abscess, and enhancing mass-like lesions (Figure 2B) can also be seen. Rarely, imaging can be normal. Most cases exhibit radiological regression, and in some instances complete normalization, following treatment. However, it is important to note that clinical outcomes might still be unfavourable despite these positive radiological changes. Imaging findings are often non-specific, with Langerhans cell histiocytosis, tuberculosis, lymphoma, and metastases presenting similarly [23].
Cavernous sinus
With only a few cases reporting this illness in the literature, cavernous sinus involvement is extremely uncommon. Imaging findings include thickening and enhancement of the cavernous sinus. They can cause cavernous sinus syndrome with the involvement of III, IV, V, and VI nerves, leading to ophthalmoplegia and facial sensory loss. Differential diagnoses include meningioma, Tolosa-Hunt syndrome, and cavernous sinus thrombophlebitis [24-26].
Vasculitis
Vascular involvement in neurosarcoidosis can either be arterial or venous predominant. Small artery perforators are affected, but large vessel involvement is uncommon. Venous predominant vasculitis occurs in the form of phlebitis along the lateral and third ventricles, involving the paraventricular veins [27,28]. The pathophysiology of vascular involvement is believed to be complex, involving factors such as infiltration of vessel walls by granulomas and dysfunction of the endothelium [13].
Deep medullary vein engorgement (DMVE) is a recently recognised cerebrovascular manifestation, which increases the risk of microhaemorrhages. On susceptibility sequences, the medullary veins are seen radially placed along the lateral ventricles (Figure 4E). The venous engorgement appears to be secondary to inflammation, causing elevated venous impedance and obliteration of the lumen. Patients with this condition are more prone to have hydrocephalus and exhibit involvement of perivascular spaces. However, this finding has not been linked with worser clinical outcomes [29]. Concomitant involvement of caudate veins is also seen in these patients. When pre-sent, dural sinus thrombosis can also cause engorgement of medullary veins [30]. The incidence of DMVE ranges from 33 to 38% [30,31]. DMVE is highly indicative of neurosarcoidosis, displaying an exceptional specificity [31].
Figure 4
Patient presented with headache and vomiting. CT brain (A) was performed, which showed acute intraparenchymal haemorrhage in the left parietal region (orange arrow). MRI brain with contrast (B-E) was performed 12 days after and showed an area of late subacute haemorrhage in the left parietal region appearing T1/T2 hyperintense with a rim of susceptibility artefact and patchy enhancement (white arrow). Susceptibility-weighted image (E) shows engorgement of deep medullary veins (yellow arrow) with linear perivascular enhancement (D) (green arrow)
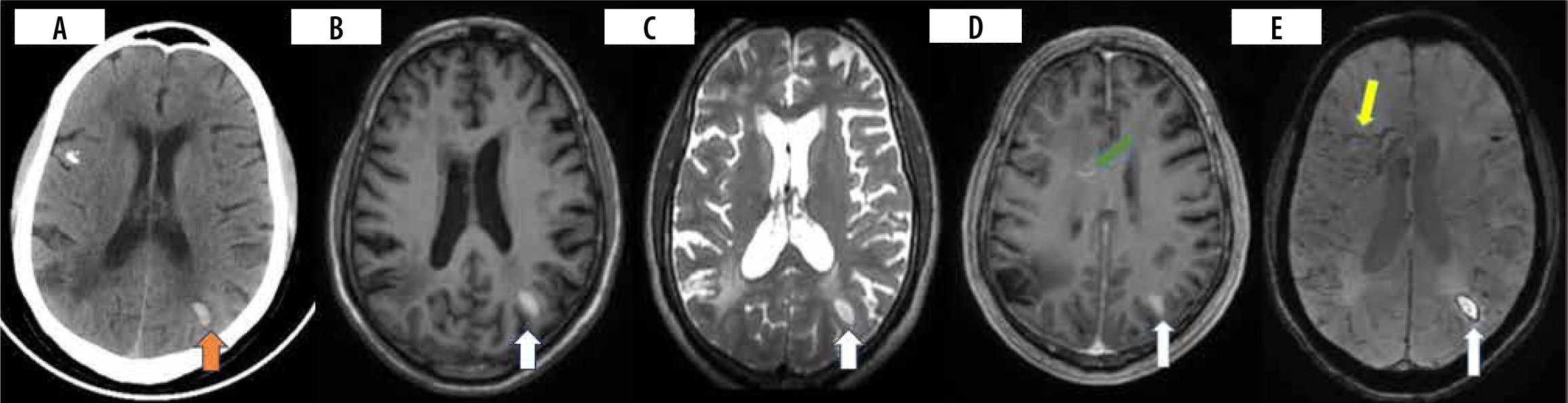
Presentation can be acute in the form of stroke, sinus thrombosis, parenchymal or sub-arachnoid haemorrhage, though rare. In many of these cases, extra neural disease is evident [13]. Intracranial haemorrhage (ICH) appears to be subcortical and lobar predominant, although perivascular enhancement is commonly seen in basal ganglia, brain stem, and spine (Figure 4A-E) [13,27]. ICH can arise as a result of both arterial and venous involvement. Both imaging and pathological observations confirm this occurrence [30,31].
Small lacunar infarcts and microhaemorrhages are frequently seen in the perforator areas, including the brainstem and basal ganglia. Tortuosity of the involved perforators might hint towards the diagnosis of neurosarcoidosis. Although major vessel involvement can be observed, ischaemic complications are rare. Many these cases have adjacent leptomeningeal inflammation. The purpose of CT/MR angiography and vessel wall imaging is to diagnose stenosis/occlusion of major arteries and perforator vasculitis, which manifests as circumferential enhancement [13,30,33]. In a cohort of 13 patients who underwent vessel wall imaging, large vessel involvement was seen in 3/13 (23%) patients while perforator vessel involvement was seen in 6/13 (46%) patients. The presence of perivascular enhancement (Figure 4D) is noted to increase the incidence of cerebrovascular events [30]. Hutto et al. assessed 11 neurosarcoidosis patients who manifested ischaemic stroke, with 4 presenting with initial ischaemic stroke and 7 experiencing strokes during the course of the disease. Small vessel involvement of lenticulostriate and pontine perforators occurred in 10/11 (90.9%), with leptomeningeal enhancement being present in all these cases [34]. Another mechanism of stroke can be due to sarcoid cardiomyopathy, causing an embolic phenomenon [31].
Spinal involvement
Spinal sarcoidosis involves a spectrum of intramedullary lesions, leptomeningitis, and pachymeningitis. It is estimated to be seen in around 1% of all sarcoidosis cases. Leptomeningeal disease appears to be the commonest, occurring in 61% of cases in which the spine is involved. Intramedullary lesions appear hypointense on T1, hyper-intense on T2, and show post-contrast nodular enhancement (Figure 5). Long-standing cases can have cord atrophy. Predilection is seen for cervicothoracic cord. Cauda equina nerve roots and conus medullaris are frequently involved. Imaging findings are frequently nonspecific, making diagnosis challenging, especially in the absence of systemic sarcoidosis. Concurrent brain involvement can be seen in up to 70% of cases [32,33].
Figure 5
Sagittal T1 and T1 + C images show long segment T2 hyperintensity (yellow arrow) with patchy post-contrast enhancement (white arrow), consistent with longitudinally extensive transverse myelitis
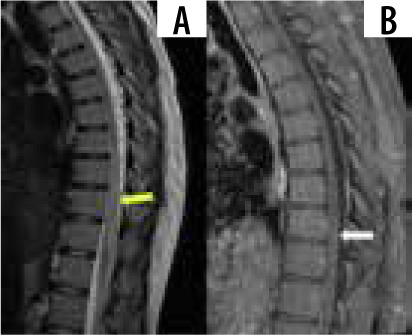
Several patterns of myelitis have been described on MRI, with longitudinally extensive transverse myelitis being the commonest, followed by short tumefactive myelitis, leptomeningeal enhancement, and anterior myelitis [34].
Sarcoid myelopathy can be easily confused with multiple sclerosis, transverse myelitis, fungal infection, glioma, or acute disseminated encephalomyelitis [12,17].
Osseous involvement
Calvarial sarcoidosis is rare, but tubular bones of the hands and feet are commonly affected. Single or multiple focal lytic lesions can be seen involving cranial convexity, skull base, orbit, and vertebral bodies. They characteristically have no peripheral sclerosis (Figure 1D). Increased uptake can be seen in bone scintigraphy. MRI findings are non-specific, showing T1 hypointensity, variable intensity on T2, and post-contrast enhancement. Lesions involving the skull base can present as cranial neuropathy. Since there are no defining imaging features in the absence of meningeal/parenchymal or extraneural involvement, multiple myeloma, metastases, and Langerhans cell histiocytosis can all be considered [35].
Peripheral nervous system
PNS involvement in neurosarcoidosis is relatively less studied compared to CNS involvement. Though cranial nerve involvement is a common presentation, involvement of the peripheral nerves is rare, occurring in about 17% of cases [6]. A variety of disorders can be observed including Guillain-Barré syndrome, polyradiculopathy, and polyneuropathy with sensory, motor, and sensori-motor features. Among these, chronic symmetrical axonal sensory-motor polyneuropathy is the commonest form. Neurosarcoidosis is an important cause of mononeuritis multiplex [3]. Large-fibre neuropathy has a better prognosis than small-fibre neuropathy [36]. Nerve ultrasound depicts enlargement, either secondary to oedema or an increase in fascicular size. Characteristically these changes are found in the distal anatomic segments of the involved nerves [37].
In many cases, muscular sarcoid remains asymptoma-tic. Pain, atrophy, and weakness can be the presenting symptoms. Elevated creatine phosphokinase (CPK), nerve conduction studies, electromyography, nuclear imaging (gallium and FDG PET), and muscle biopsy all aid in the diagnosis. When performing a biopsy, a sample from both the muscle and the nerve can be obtained, as this allows investigation of both tissues and probably increases the diagnostic yield because up to 90% of nerve biopsies can have subclinical muscle involvement. Care should be exercised because steroid therapy can also result in muscular atrophy [3,36,38].
Approach to a neurosarcoidosis patient
The Neurosarcoidosis Consortium Consensus Group (NCCC) recommends performing brain and spine MRI with and without gadolinium depending on the presentation. CSF analysis and routine workup are recommended in all cases to find corroboratory evidence and to rule out mimics. Evaluation of systemic disease should involve at least an ocular examination, high-resolution chest computed tomography, and a whole body FDG PET scan. An infectious disease workup is recommended as per the clinical history and presentation [10].
A comprehensive list of differential diagnoses of neurosarcoidosis is provided in Table 1.
Table 1
Differential diagnoses of neurosarcoidosis
Conclusions
Neurosarcoidosis poses a diagnostic and clinical challenge, given its diverse and often nonspecific manifestations. This review article provides valuable insights into the disease, highlighting the significance of recognizing its various clinical presentations and interpreting imaging findings accurately. Despite the lack of definitive imaging markers, a comprehensive diagnostic approach involving clinical assessment, advanced imaging techniques, cerebrospinal fluid analysis, and sometimes tissue biopsy is crucial for accurate diagnosis. Clinicians and radiologists must collaborate to achieve early diagnosis and tailored management strategies. With a better understanding of the complexities of neurosarcoidosis, improvement in patient outcomes and quality of care for individuals affected by this intricate disorder can be achieved.