Introduction
Neurofibromatosis type 1 (NF-1) is a common autosomal dominant genetic disorder resulting from a mutation in the NF1 gene (17q11.2 chromosome), which codes for the neurofibromin protein (regulator of RAS protooncogene) [1,2]. Individuals affected by this RASopathy show a distinct predilection to develop central nervous system (CNS) neoplasms, with the majority comprising low-grade glial tumours, including pilocytic astrocytoma (grade I) [3]. High-grade glial neoplasms (HGG) are far less common and arise more often in adults [4,5].
Utilising magnetic resonance imaging (MRI) to distinguish between low-grade and HGG in patients with NF-1 can be challenging due to limited familiarity with HGG in this patient group and the presence of occasionally equivocal imaging features. In this study, the primary focus lies on imaging features of HGG in patients afflicted with NF-1. Special attention is given to the MRI characteristics, complemented by an assessment of their clinicopathologic and molecular profiles.
Material and methods
Study design and participants
This was a retrospective analysis of histologically confirmed cases of HGG in clinically diagnosed NF-1 cases [6] over an 8-year period (2014-2022) at a tertiary cancer care centre. We included patients with baseline (treatment-naive) MRI scans available on the institutional PACS (Picture Archiving and Communication System), resulting in a sample size of 8 cases. The demographic and clinical details, including age at presentation, sex, and clinical features meeting the criteria for NF-1 diagnosis, were recorded using institutional electronic medical records (EMR).
Image acquisition, data collection and analysis
Multiparametric MRI was obtained on GE Healthcare 1.5T (Milwaukee, USA), Philips Medical Systems 1.5T (Eindhoven, Netherlands), or GE Healthcare 3T (Milwaukee, USA) MRI machines at our institute with standard brain tumour protocol (including pre- and post-contrast sequences and utilising MR-perfusion and MR-spectroscopy wherever available/applicable). Images were interpreted in conjunction by 2 radiologists with 15 and 5 years of experience, respectively. The images were reviewed on a Centricity PACS RA1000 workstation (GE Healthcare, Barrington, IL). Morphologic imaging parameters that were assessed included tumour location, size (< 3 cm, 3-5 cm, > 5 cm), number of lesions at presentation, perilesional oedema (graded as mild, moderate, or marked), necrosis, and haemorrhage. Additionally, diffusion restriction, post-contrast enhancement patterns, and leptomeningeal/ependymal spread were assessed along with relative cerebral blood flow (rCBV) values when a perfusion study was available. All scans were scrutinised for the presence of other neuroradiological stigmata of NF-1 (presence of FASIs and optic pathway glioma).
Additional histopathological (WHO grade, histology) and molecular data (ATRX status, IDH1R132H status, P53 status, H3K27M mutation) were accessed using the institutional electronic medical records (EMR). Data were analysed in MS Excel using frequency and percentage for categorical variables.
Results
General cohort characteristics (Table 1): 19 consecutive cases of clinically diagnosed NF-1 patients with HGG were identified from the neuro-oncology disease management group database from 2014-2022 (time period). Eleven cases were excluded due to the non-availability of pretreatment imaging on institutional PACS. The cohort analysed in this study consisted of 8 patients, including 6 males and 2 females, resulting in an M : F ratio of 3 : 1. The mean age of occurrence of HGG was 27.2 years (range 7-54 years). All cases met the clinical criteria for NF-1, with a positive family history of NF-1 only in one out of 8 cases.
Table 1
Demographic, clinical features
Imaging characteristics (Table 2): Of the 8 cases, 5 (62.5%) were midline neoplasms that were centred in the thalamus (n = 4/5) (Figure 1) or brainstem (n = 1/5). Cerebral lobar tumours (n = 3/8) (Figures 2 and 3) showed parietal (n = 2/3) and parieto-temporo-insular involvement (n = 1/3). One case demonstrated multifocal lesions at presentation (n = 1/8) (Figure 4), while the rest were solitary neoplasms (n = 7/8) at baseline imaging. Two out of 8 tumours (n = 2/8) were surface neoplasms, while the rest (n = 6) were parenchymal tumours.
Table 2
Magnetic resonance imaging characteristics
Figure 1
H3K27M mutant diffuse midline glioma. Axial T2, FLAIR and coronal T2WI images (A-D) show diffuse infiltrating midline tumour involving the right thalamus, medial aspect of left thalamus and extending into the midbrain. A few T2 dark areas (red arrow) are noted in the right thalamic region of the lesion, suggestive of high-grade tumour morphology. In the post-contrast study (E), the mass shows no enhancement, characteristic of midline diffuse glioma. This lesion was a histopathologically proven H3K27M mutant
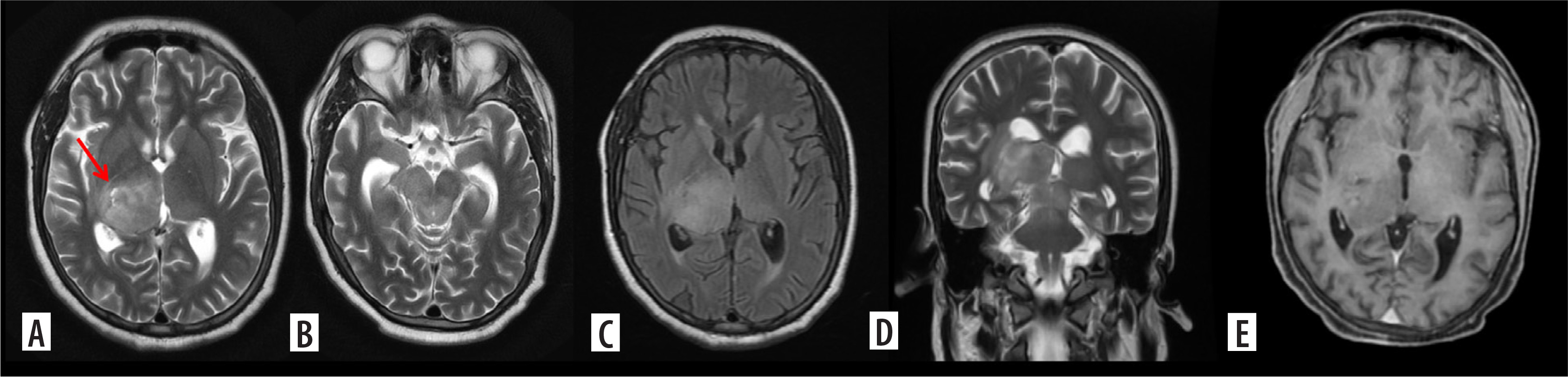
Figure 2
Glioblastoma. Axial T2, T2/FLAIR (A, B) and post contrast images (C) and coronal T2 (D) show a T2 intermediate lesion with intralesional necrosis in the right frontal lobe showing intense ring-like post contrast enhancement. Advanced magnetic resonance imaging evaluation with magnetic resonance spectroscopy shows absolute choline peak elevation with Ch:NAA 6.27 and Ch:Cr 3.42
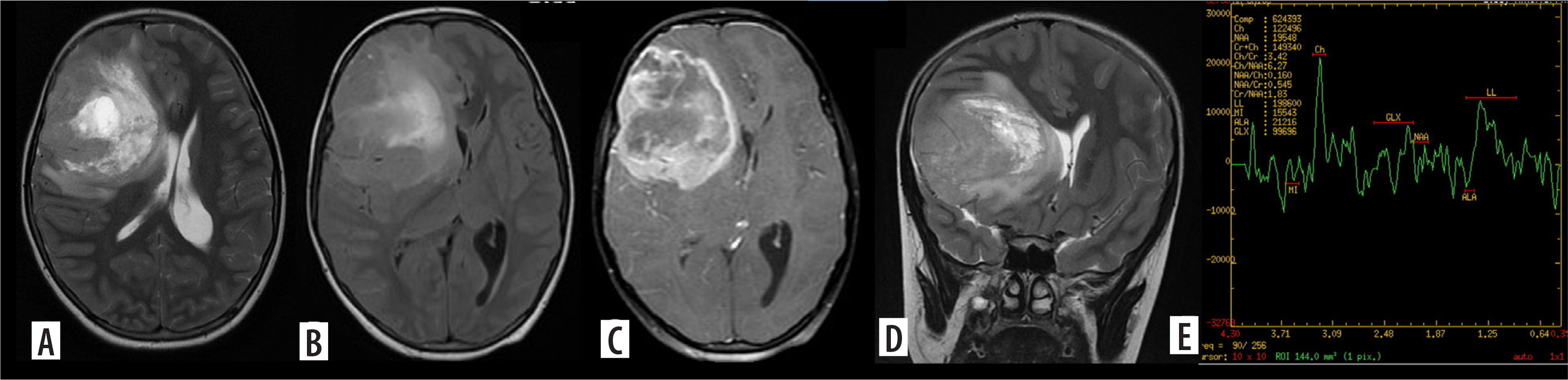
Figure 3
Gliosarcoma. Axial T2 (A) shows a cortical-based solid cystic surface-based mass in the left parietal lobe with significant perilesional edema. On axial GRE (B) the lesion shows peripheral blooming suggesting hemorrhage. On axial (C) post-contrast image, the lesion shows intense nodular enhancement and central necrotic areas
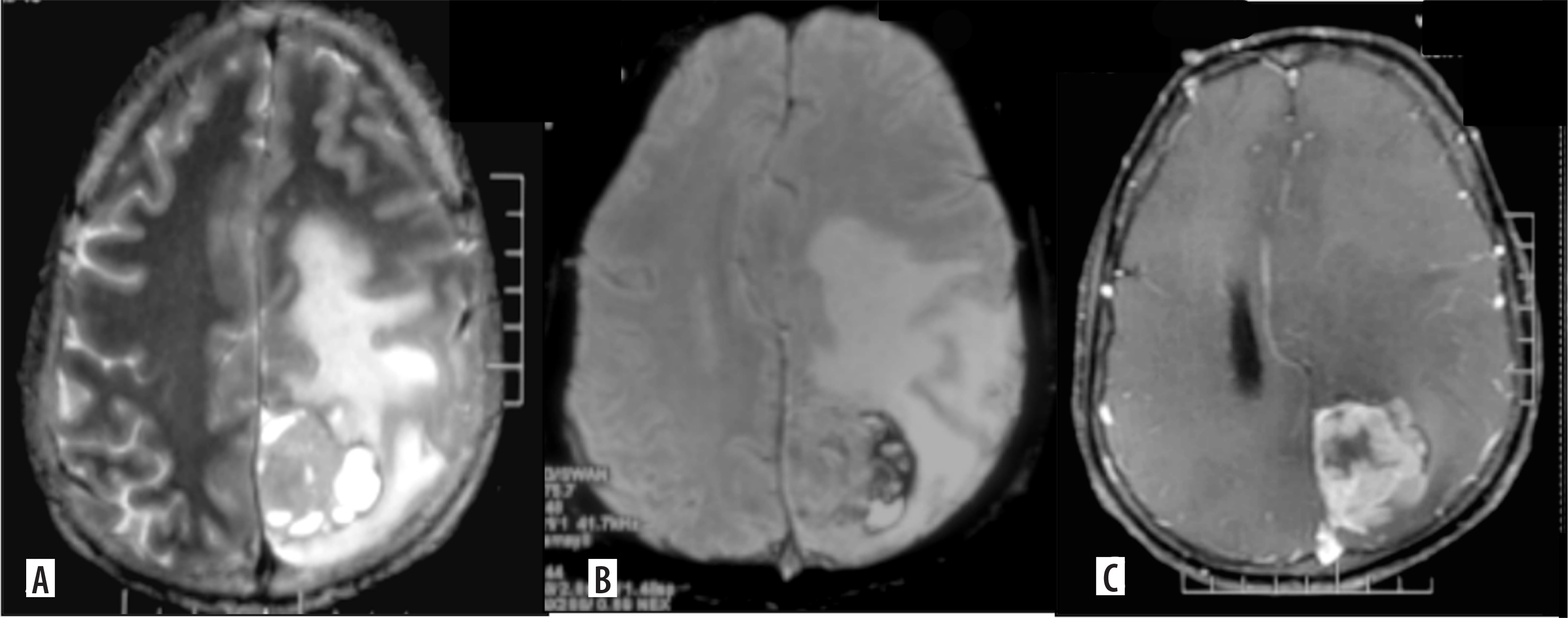
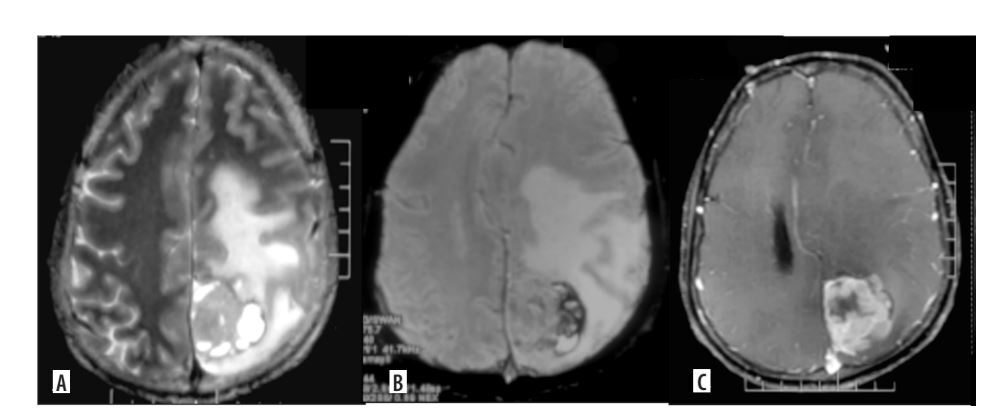
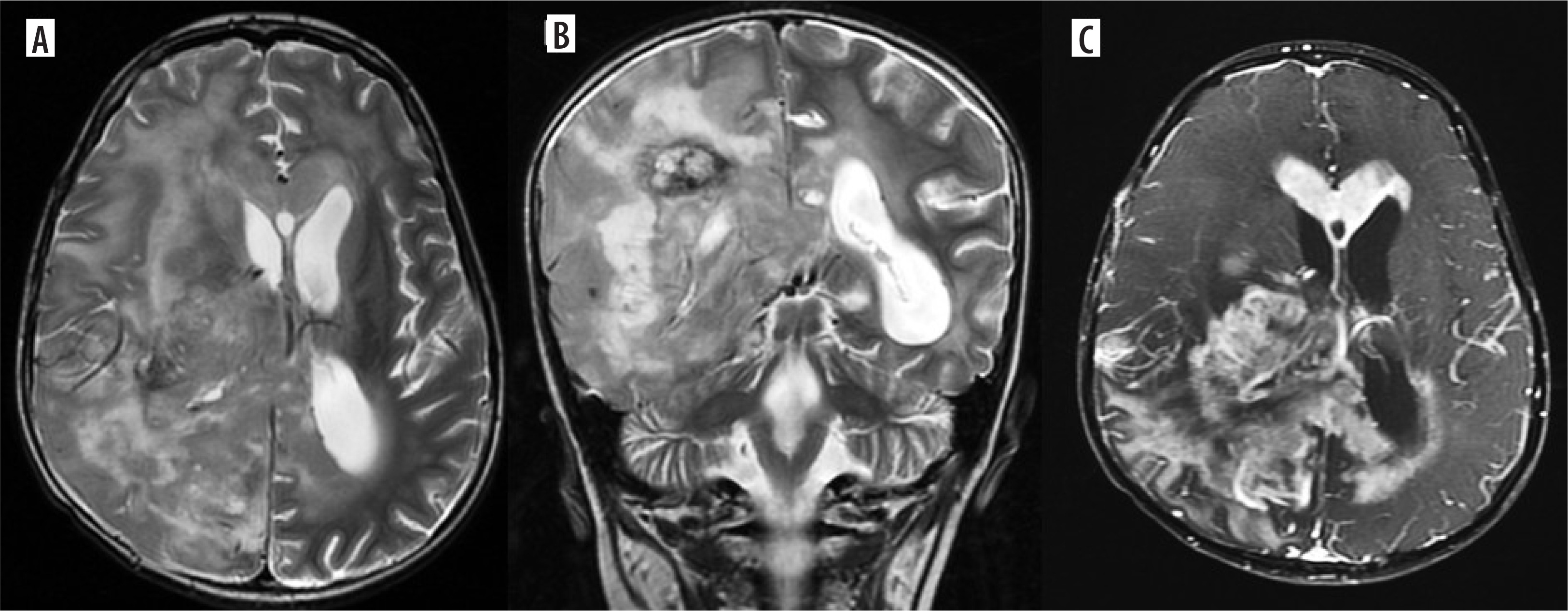
Figure 4
Multifocal glioblastoma. Axial and coronal T2 images (A, B), shows a diffusely infiltrative T2 heterogenous tumour, with heterogenous enhancement (C) in the right parieto-temporal lobe and the thalamus showing necrosis, haemorrhage and extensive surrounding non enhancing oedema. There is subependymal involvement and mild hydrocephalus, likely due to partial obstruction of the right foramen of Monro. Another discrete lesion is seen in the corpus callosum anteriorly
Most of the tumours (n = 7/8) were large in size (maximum dimension > 5 cm), with one being 3-5 cm (n = 1/8). None of the tumours were < 3 cm in the largest dimension. All lesions demonstrated some degree of perilesional oedema, mild in 4 cases (n = 4/8), moderate in 1 (n = 1/8), and marked in 2 cases (n = 2/8). Intratumoural necrosis was present in most of the tumours (n = 6/8).
The most common enhancement pattern was thick rim-like enhancement (n = 4/8), followed by predominantly nodular (n = 3/8) and predominantly solid (n = 1/8) patterns. The majority showed moderate to marked enhancement (n = 6/8), with 2 tumours only showing mild or no post-contrast enhancement (n = 2/8). Leptomeningeal dissemination at baseline scan was found in one case (n = 1/8). Perfusion study was available for 3 cases, with a mean rCBV of 3.1 : 1. Only 2 cases showed the presence of FASIs (n = 2), with no optic pathway glioma in any of the cases.
Histopathologic characteristics: Seven out of 8 cases (n = 7) had HGG that were histopathologically confirmed, with 4 glioblastoma (CNS WHO grade 4), of which one showed histology of gliosarcoma, 2 diffuse midline gliomas (DMG), H3K27M mutant (CNS WHO grade 4), and one astrocytoma, NOS (histologically CNS WHO grade 3). One of the 8 patients we examined had a midline glioma with clinicoradiological characteristics consistent with HGG without histopathological examination.
Molecular characteristics: One DMG, H3K27M mutant, and one midline glioblastoma – H3K27M-wild type on sequencing showed loss of ATRX, and they were positive for p53 and negative for IDH1R132H by immunohistochemistry. One DMG, H3K27M-mutant, showed retained ATRX and wild-type p53 staining. All other glioblastomas showed retained ATRX and were negative for IDHR132H, while p53 was positive in 2 of them. One astrocytoma NOS, negative for IDH1R132H, showed loss of ATRX and was positive for p53; however, it could not undergo further molecular evaluation.
Eight cases received standard oncological care in the form of maximum safe resection/biopsy followed by chemo-radiation (CTRT). Most of the cases (n = 6/8) showed disease progression on follow-up after initial treatment, with clinical-radiologically controlled disease in the remainder (n = 2). One case was lost to follow-up after index treatment.
Discussion
This retrospective study presents the largest reported single-centre data on imaging characteristics of HGG in NF-1. In general, the most common brain tumours in NF-1 patients are gliomas [7,8], which are found in 20% of cases, typically afflicting children as low-grade gliomas (LGG) [9,10]. Common low-grade glial neoplasms in NF-1 include pilocytic astrocytoma (WHO grade 1), which characteristically affects the optic pathway [10], and pleomorphic xanthoastrocytoma (WHO grade 2) [10-12]. Mean age at diagnosis for LGG in NF-1 is 4.5 years [10].
The mean age at presentation of NF-1-HGG in our study was 27 years, which is comparable to the mean age of 38.3 years reported by Lobbous et al. [10,13]. The age of presentation of HGGs in NF-1 appears to be lower than that of the general population [14]. In our study, 2 out of 8 cases were under the age of 18 years (ages 7 and 15). The work of Spyris et al. [13] highlights 5 rare cases of paediatric NF-1-HGG with a mean age of 7.8 years. Reported cases of glioblastomas in NF-1 have been summarised in Table 3.
Table 3
Prior cases of high-grade glial neoplasms in neurofibromatosis type 1 reported in the literature
We found that 55.5% of cases (n = 5) from our cohort were epicentred in a midline structure, of which 4 cases were adults. The average age at presentation for this subgroup was 24.6 years (range = 15 to 40 years), and 2 of them were confirmed to have H3K27M mutation, while the rest were histopathologically reported as glioblastomas or astrocytoma NOS. Cerebral HGGs arising in midline structures are characteristically aggressive tumours affecting paediatric and young adult populations [12,15]. The incidence of adult midline tumours in the general population has been reported variably but ranges between 3% and 19% [15,16]. Spyris et al. [13] discuss HGG in paediatric patients with NF-1 in their case series, in which diffuse midline glioma is the diagnosis in 4 out of 5 instances. A study by Garibotto et al. [17] concluded that cerebral HGGs arising in midline structures in paediatric patients with NF-1 is a harbinger of poorer prognosis, worse than HGGs in non-NF-1 patients. Dono et al. [16] describe characteristics of adult-type diffuse midline gliomas in their study, where 2 out of 9 diffuse midline gliomas demonstrated NF-1 gene alteration (without a statistically significant p-value). To our knowledge, no conclusive association between NF-1 and adult midline tumours has been reported in the literature. This may be due to a general paucity of literature on adult midline tumours owing to their rarity.
On imaging, large size (> 5 cm) (77.8%), intra-tumoural necrosis (77.8%), and moderate to marked perilesional oedema (55.55%) were seen in the majority of NF-1-HGG cases. Moderate to marked (77.8%) thick rim-like (55.5%) post-contrast enhancement was frequently observed and serves as an aggressive feature on imaging. These features are similar to HGGs in the general population and can aid in the prospective detection of HGGs or ‘transformed’ tumours in the NF-1 subgroup as well. An average rCBV of 3.1 : 1 was observed; however, this may have limited utility in differentiation from pilocytic astrocytomas, which are known to demonstrate hyperperfusion in their solid component with a maximum rCBV of up to 4.5 [18]. FASIs were seen only in 2/9 cases of our cohort. The synchronous presence of neuroradiologic stigmata of NF-1, such as FASIs or optic pathway glioma, appears to be unrelated to gliomagenesis [19].
Many of these high-risk imaging features were also present in one paediatric case (age 9 years) of our cohort, which also presented multifocal involvement. Although paediatric high-grade gliomas are rare in NF-1, there have been reports of aggressive HGG in this population as well [13]. It is imperative that the neuroradiologist familiarise themselves with the spectrum of paediatric NF-1-HGG so as to pick up the possible ‘atypical features’ and appropriately guide further sampling/intervention [20].
All the cases in our cohort were negative for IDH1R32H. This is consistent with previous studies, which demonstrated a lack of IDH mutations in NF-1-associated gliomas [10]. The finding that high-grade astrocytomas behave more aggressively than expected in adults with NF-1 may be partially explained by this finding. Other classic molecular features, viz. P53 positivity (71.4%, n = 5/7) and retained ATRX (n = 4/7), were also consistent with the molecular signature described by Lucas et al. [21].
We acknowledge the potential bias in the study due to its retrospective nature and relatively small sample size. While all the cases in our cohort met the clinical criteria for NF-1, none of the cases had genomic confirmation. It would be beneficial to conduct further research on these characteristics in a larger group of individuals with genomic confirmation in the future. This is especially important in the case of children, as numerous genetic variations can manifest as phenotypic NF-1.
Conclusions
We analysed the imaging features of HGGs in NF-1 patients. Younger age of presentation, larger tumour size, aggressive imaging features, and midline location should raise suspicion of higher-grade gliomas associated with this syndrome. Disease progression after treatment, in most cases, underscores the importance of adequate screening and early diagnosis.